- Research article
- Open access
- Published:
“Ask Ernö”: a self-learning tool for assignment and prediction of nuclear magnetic resonance spectra
Journal of Cheminformatics volume 8, Article number: 26 (2016)
Abstract
Background
We present “Ask Ernö”, a self-learning system for the automatic analysis of NMR spectra, consisting of integrated chemical shift assignment and prediction tools. The output of the automatic assignment component initializes and improves a database of assigned protons that is used by the chemical shift predictor. In turn, the predictions provided by the latter facilitate improvement of the assignment process. Iteration on these steps allows Ask Ernö to improve its ability to assign and predict spectra without any prior knowledge or assistance from human experts.
Results
This concept was tested by training such a system with a dataset of 2341 molecules and their 1H-NMR spectra, and evaluating the accuracy of chemical shift predictions on a test set of 298 partially assigned molecules (2007 assigned protons). After 10 iterations, Ask Ernö was able to decrease its prediction error by 17 %, reaching an average error of 0.265 ppm. Over 60 % of the test chemical shifts were predicted within 0.2 ppm, while only 5 % still presented a prediction error of more than 1 ppm.
Conclusions
Ask Ernö introduces an innovative approach to automatic NMR analysis that constantly learns and improves when provided with new data. Furthermore, it completely avoids the need for manually assigned spectra. This system has the potential to be turned into a fully autonomous tool able to compete with the best alternatives currently available.

Self-learning loop. Any progress in the prediction (forward problem) will improve the assignment ability (reverse problem) and vice versa.
Background
The automation of chemical analysis by nuclear magnetic resonance (NMR) spins around two problems: the forward problem of predicting the NMR spectra of a given molecule, and the inverse problem of elucidating the molecular structure that generates a given experimental spectrum. The forward problem is solved, in principle, by quantum mechanics: molecular structure determines a unique Hamiltonian from which all measurable NMR parameters can be computed. However, this solution is impractical in most cases of interest. First, the ab initio prediction on a personal computer of the NMR parameters (chemical shifts and scalar couplings) for a small molecule takes at least as long as the actual experiments. Second, an isolated molecule is actually a very poor model for a real NMR spin-system in solution, as it ignores solvent effects and the existence of multiple conformations. Accounting for these issues, if possible, would imply even longer calculations. Thus, ab initio prediction of NMR parameters is not a suitable approach for automatic analysis of NMR data.
In practice, the forward problem of NMR prediction is handled by semi-empirical methods based on previous knowledge about typical chemical shifts. Indeed, several commercial packages exist that perform NMR prediction based on models adjusted to large databases of observed chemical shifts [1–9]. To build such databases nuclei must be assigned to observed chemical shifts, a task that concerns the much more challenging inverse problem. Furthermore, predicted chemical shifts play an important role in the assignment process as well. The two problems are thus strongly related, a fact that poses an important limitation to the automation of NMR analysis. This reflects in existing computational tools for NMR elucidation and assignment: either they are not fully-automatic, requiring preliminary analysis by the user [10, 11], or resort to chemical shift predictions [10, 12–18] that rely on databases of spectra assigned ‘manually’ by trained experts. Regardless of the approach, a significant amount of labour is involved that is certainly not devoid of human errors.
We can turn this issue around by noting that the strong relation between the forward and inverse problems means that progress in one direction improves the other [19]. Indeed, successful assignment of a spectrum generates information that can enrich the database used by an NMR predictor. In the opposite direction, more accurate and reliable chemical shift predictions facilitate rejection of non-viable assignments.
This relation then allows to devise a fully automatic assignment and prediction system that progressively improves its capabilities. Learning ex nihilo, however, requires an automatic assignment method that is able to assign several spectra without resorting to chemical shift prediction. We developed such method elsewhere [20] and use it here to create Ask Ernö, Footnote 1 a fully autonomous assignment—prediction program.
Methods
The concept behind Ask Ernö is summarized in Fig. 1. Automatic assignment of a nucleus in a molecule associates a substructure (the nucleus and its surroundings) with an observed chemical shift. This information can be stored in a database and used to predict chemical shifts. As the database grows, the accuracy of the predictor improves. The improved predictor, in turn, provides better chemical shift constraints to be used in subsequent assignments. Ask Ernö learns by running repeated assignment cycles on a given training set, using each new assignment to improve its predictions in the next cycle.

The logic behind Ask Ernö. The automatic assignment of nuclei to their signals (right) produces entries to a database (mid) for chemical shift prediction (left). Predicted chemical shifts in turn provide further restrictions for assignment. Ask Ernö is trained by repeatedly looping on this assignment-prediction cycle
Ask Ernö was implemented as a proof of concept rather than a full-fleshed assignment and prediction tool. For this reason, it was designed with small molecules in mind and tested only with 1H NMR data.
Chemical shift prediction
Each entry in the database for chemical shift prediction consists of two terms:
-
F: a molecular fragment around a proton, comprising the substructure spanned by all atoms up to n bonds from it, with n ∊ {1, 2,…}. We refer to this fragment as the n-sphere around the proton and to n as its radius or size (see Fig. 2). These fragments are stored as Hierarchically Ordered Spherical description of Environment (HOSE) codes [22].
Fig. 2 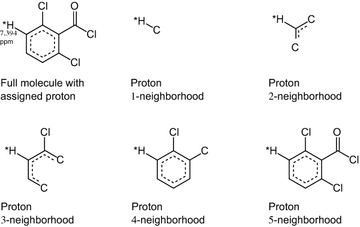
n-spheres with radius 1–5 of a proton assigned to a chemical shift of 7.394 ppm. Dotted curves indicate aromatic bonds
-
δ: an observed chemical shift value for the proton.

These registers are generated by automatic assignment of experimental spectra (see Learning for details). Since the same fragment F may be observed and assigned in different molecules, multiple entries may exist for each fragment.
Predictions were done using the HOSE-based methodology developed in CSEARCH for 13C-NMR [24] and later implemented in Modgraph NMRPredict [25]. For 1H-NMR it works as follows: the predictor spans the n-sphere of radius n max around the proton of interest, encodes the resulting fragment as a HOSE code, and queries it on the database. If the query is successful, the median \(\underline{\delta }\) over all matches is returned as the predicted chemical shift, with an uncertainty \(\varepsilon\) equal to their standard deviation. If no matching entries are found, a new query is sent for the n-sphere of radius n max − 1 around the proton and so on, until a successful match is found or the radius of the sphere is below n min. In the latter case the predictor returns a failed status.
Assignment
We used an automatic assignment method previously described in [20] that performs fully automatic peak-picking and assignment of chemical shifts based on peak integrals (signal intensity), 2D spin–spin correlations, and chemical shifts. The assignment routine uses a symmetry-constrained branch and bound optimization that achieves a thorough exploration of the whole solution space. The result is a list of of assignments, ranked according to how well they fit the observed data. This automatic assigner has been shown to yield good results even if no chemical shift data is provided, which is of great importance for the present development.
Since we only used 1H-NMR spectra, assignments were performed exclusively on the basis of integration and chemical shift data. The auto-assigner was configured to seek for assignments that perfectly matched the observed integrals (rounded to the closest integer), and that matched the predicted and observed chemical shifts (when available) with an error no greater than 3 times the prediction’s uncertainty at the current iteration. For this purpose, the uncertainty was estimated as the standard deviation of the sample of observed chemical shifts on which the prediction is based (see Chemical shift prediction above), multiplied by the following factor:
where n is the size of the sample and I is the index of the current iteration (see Learning below). This factor contributes significantly to controlling the propagation of error, since the standard deviation is a poor estimator of the uncertainty for small n or I. Furthermore, for predictions based on less than two matches the allowed chemical shift error was set to the maximum of 20 ppm, considering that no reasonable estimation of uncertainty is possible in that situation.
Learning
The learning algorithm is based on a self-organizing map and consists of a recursive cycle on the training dataset, which is repeated until nothing new is learnt. The first learning iteration starts by running the automatic assignment algorithm without taking chemical shifts into account. We refer to it as iteration 0. Redundancy (e.g. multiple occurrences of methyl groups) is expected so that several possible assignments may be found for any given molecule; this is particularly true when no spin–spin correlation data (2D NMR experiments) is available. Though a unique solution is unlikely, it is often possible to find some nuclei—chemical shift dyads that are present in all assignments computed for a molecule and that can thus be assumed to be correct (see Table 1). These dyads are learnt by creating database entries for the corresponding n-spheres, with n = n min,…, n max (see Chemical shift prediction above).
Completing this process on all molecules of the training set finishes iteration 0. The system then proceeds with iterations 1, 2, etc., in which newly learnt chemical shifts are used as additional restrictions for subsequent assignments. Database entries are batch-generated, that is, chemical shifts learnt in one cycle are only available starting from the next one. We found in preliminary tests that this approach slows down the learning process but yields better results than the “on-line” approach. Learning continues until two consecutive iterations yield no improvement.
Experimental
Ask Ernö was implemented in Java (automatic assigner), MySQL (prediction database) and JavaScript (chemical shift predictor, self-learning loop and integration of the system’s components). The project is open source and available on GitHub [23], along with links to the data used for training and testing. A web service is available at https://www.cheminfo.org/flavor/askerno/index.html for anyone willing to evaluate the system.
The data used for the evaluation consisted of 2639 molecules along with their experimental 1H-NMR spectra. Examples of these spectra are included as Additional file 1. The dataset was assembled by random sampling from the Maybridge catalogue (2198 selected registers) and from our own library (441 selected registers). Data was split in a training set (2341 molecules, Additional file 2) and a test set (298 molecules, Additional file 3). No assignment information was provided along with the training set. Spectra in the test set were manually assigned to determine the reference experimental chemical shift values for the calculation of prediction error. Not all protons in the set were assigned. Most remarkably, labile protons were avoided considering that they are known to pose challenges to the components of Ask Ernö [20] and that we intended to evaluate the potential and issues of the self-learning loop rather than those of its components. Overall, 2007 assigned protons were used to test Ask Ernö’s predictions.
Ten iterations of training were run, with n max = 4 and n min = 2. At the end of each iteration, chemical shifts for the test molecules were predicted and compared with the observed values.
Results and discussion
Figure 3 shows the evolution of the correlation between predicted and observed chemical shifts through 10 iterations. It can be seen that predictions oscillate from one iteration to the other as they converge towards the observed value (diagonal). Indeed, at the last iteration, most predictions accumulate close to the diagonal, though a few large errors persist.

Correlation between observed and predicted chemical shift values for the test molecules at each iteration of the training loop
To get a more detailed picture of Ask Ernö’s performance and learning process we looked at three indicators: prediction error, prediction uncertainty, and the fraction of chemical shifts from the test set that could be predicted.
Prediction error
The overall prediction error is expected to decrease as the system iterates, and final errors to be the lowest possible. Figure 4 (top) shows the evolution of the average error across the iterations for n min = 2, 3, 4. It is found that larger n min values yield lower errors, but also that it improves less through each iteration (slower learning). Indeed, a larger sphere radius gives a better representation of the magnetic environment of the proton of interest, producing a more accurate prediction that can hardly be improved. For smaller fragments the distribution of observed chemical shifts is wider, so there is more room for improvement. Thus, as the system iterates and the database of fragments grows, the average chemical shift of matching fragments moves closer to the true mean of the full distribution, lowering the average prediction error. For n min = 2 this error decreased by 17 % across 10 cycles, for a final value of 0.265 ppm.

Evolution during the training loop of prediction error (top), prediction uncertainty (middle) and fraction of predicted chemical shifts (bottom) for the test molecules
Since the average error can be dominated by a few predictions with large errors, the cumulative error distributions were plotted (Fig. 5). It can be seen that larger n min values yield a higher number of accurate predictions (<0.2 ppm) and fewer predictions with high error (>1 ppm). Also, the number of accurate predictions grows faster with larger n min (12 % at n min = 4 vs. 4 % at n min = 2), while the number of less accurate predictions reduces more slowly (<1 % for n min = 4 vs. 4 % at n min = 2). This is consistent with the observed behavior of the average error and again is explained by the naturally higher accuracy of predictions achieved with larger n-spheres. In the end, with n min = 2, over 60 % of the tested chemical shifts were predicted with less than 0.2 ppm error, and only 5 % of them were found with error exceeding 1 ppm.

Evolution of the cumulative error distributions during training. The fraction of predictions is given relative to the total number of protons in the test set for which chemical shift can be predicted (2007 protons in total). To generate these curves, the set of chemical shift prediction errors was split into 100 bins of 0.01 ppm, plus a last bin containing predictions with an error equal or greater than 1 ppm. This last bin being larger explains the sudden increase observed at the end of the curves
Prediction uncertainty
In Ask Ernö, the uncertainty of a prediction is associated with the standard error of the distribution of chemical shifts of matching fragments (see Methods, Chemical shift prediction). While the prediction error validates the results against an external reference (the correct chemical shifts), the uncertainty provides an internal validation. It is expected that as the system learns it gives predictions with lower uncertainty.
Figure 4 (mid) presents the evolution of this statistic through the training. It can be seen that the uncertainty quickly decreases, reaching a limit value. Both the rate and limit value are related to n min : the smaller n min the faster the uncertainty decreases and the lower it reaches (0.23 ppm for n min = 2 and < 0.1 ppm for n min = 4).
Note that this limit is nothing but the standard deviation of the distribution of chemical shifts on the population of all possible n-spheres for the corresponding n min . This allows for an interesting interpretation of the limit uncertainty as the theoretical best that Ask Ernö can achieve. Noting how the final average error in Fig. 4 (top) is above the limit uncertainty in Fig. 4 (mid), we conjecture that Ask Ernö’’s accuracy can still be improved by around 13 % through further training with more data.
Amount of predicted chemical shifts
For a chemical shift to be predicted, it is necessary that a matching substructure is found in the database. The fraction of chemical shifts from the test set that can be predicted then constitutes a third descriptor of learning. Figure 4 (bottom) shows that though larger n-spheres provide better predictions, they only cover around half of the test problems (54 % for n min = 4 at the end of learning). Including predictions with n min = 3 and n min = 2 allows for a major leap in coverage, up to 85 and 99 %. It is clear that no significant improvement can be gained by considering 1-spheres.
It is worth noting that the fraction of predictions with larger n-spheres increases by 13 % during training. This is pivotal to Ask Ernö’s performance: as its database grows, larger n-sphere matches becomes possible, which translates into a higher number of more accurate predictions.
Sources of error
Ask Ernö is particularly prone to errors when working with structures underrepresented in the training set. For instance, consider a prediction based on a small fragment that is present in numerous molecules of the training set. Since this small fragment is unable to properly account for all relevant interactions, it is associated with a broad range of chemical shifts and the uncertainty of the prediction is very high. Although such fragments are only used until a bigger match is found, no better match will ever be found for underrepresented fragments. In other words, Ask Ernö can’t learn to correctly predict spin systems that are not properly represented in the training set. The situation just described is reflected in the large lines of vertically aligned points, observed in Fig. 6. Most mistakes are located along these vertical series of points, proving that this was the main source of error in the test.

Correlation between observed and predicted chemical shift values after learning for different sphere radius (iteration 9)
Other recurring mistakes can be related to underrepresented structures. For instance, the biggest errors for predictions based on 4-spheres (see Fig. 6, bottom) arose when the query returned a single matching fragment. In these cases the maximum uncertainty (20 ppm) given by the assigner to predictions based in less than 3 fragments allows for the propagation of an error that in principle should be rectified by new observations, but remains due to lack of the necessary data.
Detailed examples are given in the Additional file 4.
Conclusions
The reduction in error and uncertainty and the increase in the amount of predictions proves that Ask Ernö is indeed improving its prediction capabilities as it iterates on the assignment-prediction cycle. After 10 iterations using a set of 2341 assignment problems, Ask Ernö was able to predict the chemical shifts of protons in a set of 298 molecules with an average error of no more than 0.265 ppm. At least 60 % of the chemical shifts were predicted with an error of less than 0.2 ppm. These are very promising results, especially for such a basic implementation of the concept.
It must be emphasized that Ask Ernö developed this capability fully autonomously: at no point it was fed with the fruits of the labour of human experts. The learning process of Ask Ernö is akin to that of a newcomer to the realm of NMR analysis, who is told the basic rules of assignment and through experience and induction develops his own NMR tables.
As expected, larger n-spheres provide better but fewer predictions. Furthermore, it was found that most errors occurred for underrepresented molecules when forcing highly uncertain predictions based on smaller fragments. For these reasons, it is expected that with more data the database could grow to a point where any query would match a large n-sphere. Thus, though the system currently tops at an average error of 0.265 ppm, the limit of <0.1 ppm error could be reached with enough data. Further improvements to this limit would require taking into account other experimental parameters such as solvent, concentration and temperature of acquisition, as major source of experimental errors.
Based on the results presented here, we expect to develop Ask Ernö into a state-of-the-art tool for automatic NMR analysis in the near future. Current efforts are focused in reforming the estimator of uncertainty in order to enhance the system’s capability to rectify its mistakes as it iterates. Correlation data from 2D experiments should also lead to significant improvement, when available.
Availability of data and materials
The source code used in this work is available in the github repository https://github.com/cheminfo/autolearning. The dataset supporting the conclusions of this article is available in the github repository https://github.com/cheminfo/autolearning. The molecules in the dataset supporting the conclusions of this article are also included within the article (and its Additional file 2, Additional file 3) as sdf files.
Notes
In reference to Ernö Pretsch and his classic book compiling thoroughly the necessary information for humans to assign spectra [21].
Abbreviations
- NMR:
-
nuclear magnetic resonance
- HOSE:
-
hierarchically ordered spherical description of environment
References
ACD/HNMR Predictor v.9.0, Advanced Chemistry Development, Inc., 110 Yonge Street, 14th floor, Toronto, Ontario, Canada M5C1T4
Binev Y, Marques MM, Aires-de-Sousa J (2007) Prediction of 1H NMR coupling constants with associative neural networks trained for chemical shifts. J Chem Inf Model 47(6):2089–2097
Aires-de-Sousa J, Hemmer M, Gasteiger J (2002) Prediction of 1H NMR chemical shifts using neural networks. Anal Chem 74(1):80–90
Binev Y, Corvo M, Aires-de-Sousa J (2004) The impact of available experimental data on the prediction of 1H NMR chemical shifts by neural networks. J Chem Inf Comput Sci 44(3):946–949
Binev Y, Aires-de-Sousa J (2004) Structure-based predictions of 1H NMR chemical shifts using feed-forward neural networks. J Chem Inf Comput Sci 44(3):940–945
Binev Y, Marques MM, Aires-de-Sousa J (2007) Prediction of 1H NMR coupling constants with associative neural networks trained for chemical shifts. J Chem Inf Model 47(6):2089–2097
SPINUS WEB http://www2.ccc.uni-erlangen.de/services/spinus/. Accessed Feb 2016
Modgraph NMR Prediction http://www.modgraph.co.uk/product_nmr.htm. Accessed Feb 2016
ChemDraw 15.0 http://www.cambridgesoft.com/Ensemble_for_Chemistry/ChemDraw/. Accessed Feb 2016
Elyashberg ME, Williams AJ, Martin GE (2008) Computer-assisted structure verification and elucidation tools in NMR-based structure elucidation. Prog Nucl Magn Reson Spectrosc 53:1–104
Plainchont B, Emerenciano VP, Nuzillard J-M (2013) Recent advances in the structure elucidation of small organic molecules by the LSD software. Magn Reson Chem 51:447–453
Griffiths L, Beeley HH, Horton R (2008) Towards the automatic analysis of NMR spectra: part 7. Assignment of 1H by employing both 1H and 1H/13C correlation spectra. Magn Reson Chem 46:818–827
Golotvin SS, Vodopianov E, Pol R, Lefebvre BA, Williams AJ, Rutkowse RD, Spitzer TD (2007) Automated structure verification based on a combination of 1D 1H-NMR and 2D 1H-13C HSQC spectra. Magn Reson Chem 45:803–813
Cobas C, Seoane F, Vaz E, Bernstein MA, Dominguez S, Pérez M, Sýkora S (2013) Automatic assignment of 1H-NMR spectra of small molecules. Magn Reson Chem 51:649–654
Masui H, Hong H (2006) Spec2D: a structure elucidation system based on 1H-NMR and H-H COSY spectra in organic chemistry. J Chem Inf Model 46:775–787
Molodtsov S, Elyashberg ME, Blinov KA, Williams AJ, Martirosian EE, Martin GE, Lefebvre B (2004) Structure elucidation from 2D NMR spectra using the StructEluc expert system: detection and removal of contradictions in the data. J Chem Inf Comput Sci 44:1737–1751
Steinbeck C, Kuhn S (2004) NMRShiftDB - compound identification and structure elucidation support through a free community-built web database. Phytochemistry 65:2711–2717
Meiler J, Will M (2001) Automated structure elucidation of organic molecules from 13C-NMR spectra using genetic algorithms and neural networks. J Chem Inf Model 41:1535–1546
Tarantola A (2005) Inverse problem theory and methods for model parameter estimation. SIAM, Philadelphia
Castillo AM, Bernal A, Patiny L, Wist J (2015) Fully automatic assignment of small molecules’ NMR spectra without relying on chemical shift predictions. Magn Reson Chem 53:603–611. doi:10.1002/mrc.4272
Pretsch E, Bühlmann P, Affolter C (2000) Structure determination of organic compounds. Springer, Berlin
Bremser W (1978) HOSE—a novel substructure code. Anal Chim Acta 103:355–365
Ask Ernö in Github https://github.com/cheminfo/autolearning. Accessed Feb 2016
Kalchhauser H, Robien W (1985) CSEARCH: a computer program for identification of organic compounds and fully automated assignment of carbon-13 nuclear magnetic resonance spectra. J Chem Inf Comput Sci 25:103–108. doi:10.1021/ci00046a010
Modgraph C13 NMR prediction http://www.modgraph.co.uk/product_nmr_HOSE.htm Accessed Feb 2016
Authors’ contributions
LP devised the basic concept of a self-learning assigner and predictor. AMC developed the implementation, supervised by JW and LP. AB drafted the manuscript with the help of JW. JW and AB achieved all illustrations jointly. RD strongly supported the manual revision of assignments and tracking down of issues with the peak-picking routine. All authors participated in the design and interpretation of the test and in the redaction of the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Funding
AMC thanks the program Créditos-Beca Francisco José de Caldas, convocatoria 528 of Colciencias for funding.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Castillo, A.M., Bernal, A., Dieden, R. et al. “Ask Ernö”: a self-learning tool for assignment and prediction of nuclear magnetic resonance spectra. J Cheminform 8, 26 (2016). https://doi.org/10.1186/s13321-016-0134-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13321-016-0134-6