- Research article
- Open access
- Published:
Efficient enumeration of monocyclic chemical graphs with given path frequencies
Journal of Cheminformatics volume 6, Article number: 31 (2014)
Abstract
Background
The enumeration of chemical graphs (molecular graphs) satisfying given constraints is one of the fundamental problems in chemoinformatics and bioinformatics because it leads to a variety of useful applications including structure determination and development of novel chemical compounds.
Results
We consider the problem of enumerating chemical graphs with monocyclic structure (a graph structure that contains exactly one cycle) from a given set of feature vectors, where a feature vector represents the frequency of the prescribed paths in a chemical compound to be constructed and the set is specified by a pair of upper and lower feature vectors. To enumerate all tree-like (acyclic) chemical graphs from a given set of feature vectors, Shimizu et al. and Suzuki et al. proposed efficient branch-and-bound algorithms based on a fast tree enumeration algorithm. In this study, we devise a novel method for extending these algorithms to enumeration of chemical graphs with monocyclic structure by designing a fast algorithm for testing uniqueness. The results of computational experiments reveal that the computational efficiency of the new algorithm is as good as those for enumeration of tree-like chemical compounds.
Conclusions
We succeed in expanding the class of chemical graphs that are able to be enumerated efficiently.
Introduction
The enumeration of chemical structures satisfying given constraints is an important topic in chemoinformatics [1–3]. Applications of the enumeration of chemical structures include structure determination using mass-spectrum and/or NMR-spectrum [4, 5], virtual exploration of the chemical universe [6, 7], reconstruction of molecular structures from their signatures [8, 9], and classification of chemical compounds [10]. The enumeration problem is also important for development of novel chemical compounds because virtual exploration of chemical universe and reconstruction of molecular structures from their signatures are considered to be important elementary technologies. The enumeration of chemical structures has a long history. Cayley [11] considered the enumeration of structural isomers of alkanes in the 19th century. The seminal work of Pólya on counting the number of isomers using group theory is also famous [12].
In this paper, we consider the problem of enumerating chemical structures having monocyclic graph structures satisfying a given constraint, where a monocyclic graph is an undirected connected graph containing exactly one cycle (a graph is connected if there exists a path connecting every pair of vertices), and a constraint is given in the form of a set of feature vectors (i.e., a set of descriptors). We assume that each feature vector specifies the number of occurrences of each labeled path of length up to a given constant K, where a labeled path is an alternating sequence of atom names and bond types (see Figure 1 for an example of a feature vector). We also assume that a set of feature vectors is given by specifying the minimum and maximum numbers of occurrences of each labeled path. We develop an efficient algorithm for this enumeration problem by extending existing algorithms [13, 14] for enumerating tree-like chemical structures (i.e., chemical structures without cycles). In this extension, some novel concepts are introduced and rigorous mathematical analysis is performed in orer to guarantee the correctness of the algorithm.

Example of a chemical structure and its feature vector. In this example, a feature vector consists of the number of occurrences of each atom type and each bond type (e.g., C2C denote the double bond between two carbon atoms). Note that the entry of C1C is 8 because each bond is counted for both directions. Since there may exist multiple structures with the same feature vector, enumeration of such structures is required.
In order to verify the computational efficiency of our proposed algorithm, we perform computational experiments using a set of some chemical compounds from the KEGG LIGAND database [15]. The results suggest that the proposed algorithm enumerates chemical structures having monocyclic graph structures as nearly efficiently as tree-like chemical graphs have been enumerated.
The rest of this paper is organized as follows. First, we review some mathematical definitions and give a formal definition of the enumeration problem for chemical structures with monocyclic graph structures. Next, we review background and related work. Then, we present the algorithm and the results of computational experiments. Finally, we conclude with future work. Mathematical proofs, pseudocodes for the algorithm, and some details on computational experiments are given in Additional file 1.
Preliminaries and problem formulation
This section reviews some basic definitions on graphs and formalizes the problem to be addressed in this work. Before providing formal descriptions, we briefly explain the problem definition using an example in Figure 2.

Example of a Σ - colored multi-graph G and its feature vector f 1 ( G ). G represents a hydrogen-suppressed chemical graph, where deg(v;G)<val(c(v)) holds for some vertex v∈V(G). Since each path is counted for both directions, the entry for C2C is not one, but two.
Our basic problem is to enumerate all chemical structures each of which is consistent with a given feature vector and the valence condition. Each coordinate of a feature represents the number of occurrences of vertex- and edge-labeled paths. In order to keep the size of a feature vector moderate, we restrict the length of paths to be no greater than a constant K. In the example of Figure 2, we consider paths of lengths 0 and 1, where a path of length 0 corresponds to a single atom and a path of length 1 corresponds to a bond including its endpoint atoms. For example, the columns O, N, and C of feature vector f1(G) mean that each target structure must contain exactly one oxygen, two nitrogen, and three carbon atoms, respectively. The columns N1O, N1C, and C2C mean that each target structure must contain exactly one single bond connecting N and O, two single bonds connecting C and N, and one double bond connecting C and C. It should be noted that one single bond connecting N and O is counted by both O1N and N1O. Then, the chemical structure G is consistent with f1(G). However, another chemical structure may be consistent with a given feature vector. For example, the feature vector remains the same even if the double bond (along with the branching carbon atom) is moved into the backbone chain. Therefore, it is desirable to enumerate all chemical structures consistent with a given feature vector and the valence condition (specified by val(…)). On the other hand, there may not exist any consistent chemical structure if K is large; thus it may not be appropriate to uniquely specify a feature vector. Therefore, we assume in our target problem that upper and lower bounds of the number of occurrences of each labeled path are given as shown in Figure 3.

Example of an input of EULF and part of its output. The input includes upper and lower feature vectors, and the output includes multi-trees G1 and G2 and a 1-augmented tree G3.
A multi-graph is a graph that can have multiple edges between the same pair of vertices, where vertices correspond to atoms and multi-edges correspond to double and triple bonds in chemical compounds. We call a connected multi-graph a k-augmented tree if the number of adjacent vertex pairs (i.e., vertex pairs connected by edges) minus the number of vertices is k−1 (hence a multi-tree is a 0-augmented tree). That is, a k-augmented tree is a graph obtained by adding edges to k different pairs of nonadjacent vertices in a multi-tree (see Figure 4). The problem considered in this paper is to enumerate all 1-augmented trees satisfying specified upper and lower bound conditions on feature vectors. In the following, we provide the mathematical definition of the problem. We assume that readers have some familiarity with basic concepts in graph theory. For those who are not familiar with graph theory, we suggest referring to an appropriate textbook (e.g., [16]). Readers not interested in mathematical details can skip this part.

Examples of k -augmented trees. A k-augmented tree is obtained by adding k edges to a multi-tree, where a multi-tree is a tree with multiple bonds (precisely, multiple edges between the same pair(s) of vertices).
A graph is defined to be an ordered pair (V,E) of a finite set V of vertices and a finite set E of edges, where an edge is an unordered pair of distinct vertices (thus no self-loop exists), where an edge with two end-vertices u and v is denoted by uv. A graph is called a multi-graph when E is not necessarily composed of distinct pairs of vertices (thus multiple edges are allowed in a multi-graph and E is no longer a set, but a multi-set), and is called a simple graph if no multiple edges are allowed. The multiplicity (the number of multiple edges) between two vertices u and v is denoted by m(u,v). An edge in a multi-graph G is called simple if its multiplicity in G is one. We denote the vertex set and edge set of a graph G by V(G) and E(G), respectively. For a vertex v in a multi-graph G, let deg(v;G) denote the number of edges incident to vertex v (i.e., degree of v). In this paper, a cycle is a closed path with a length at least three (two edges with the same endvertices are not treated as a cycle), and a connected multi-graph (resp., simple graph) with no cycle is a multi-tree (resp., simple tree). A k-augmented tree is a connected multi-graph such that the number of adjacent vertex pairs (i.e., vertex pairs connected by edges) minus the number of vertices is k−1. For two vertices u and v in a multi-graph G, let G+u v denote the multi-graph obtained by adding a new edge uv to G; when u v∈E(G), let G−u v denote the multi-graph obtained by removing uv from G. Let denote the set of nonnegative integers, and let Σ be a set of colors, which correspond to chemical elements such as H, O and C. Let each color c∈Σ be associated with a valence. A multi-graph G is said to be Σ−colored if each vertex v has a color c(v)∈Σ. Chemical compounds can be viewed as Σ-colored, self-loopless connected multi-graphs, where vertices and colors represent atoms and elements, respectively.
Let be a prescribed integer, which corresponds to the maximum multiplicity of chemical graphs, and Σk,d denote the set of all alternating sequences (c0,m1,c1,…,m k ,c k ) consisting of colors c0,c1,…,c k ∈Σ and m1,m2,…,m k ∈{1,2,…,d}. We denote the union of Σ0,d, Σ1,d, Σ2,d,…,Σk,d by Σ≤k,d. Let be the set of all mappings g from Σ≤k,d to , i.e., .
For a path P=(v0,m1,v1,…,m k ,v k ) such that V(P)={v0,v1,…,v k }, E(P)={v0v1,v1v2,…,vk−1v k }, and m i =m(vi−1,v i ) is the multiplicity of edge vi−1v i , the length of P is defined to be k=|V(P)|−1, and the color sequence c(P) of P is defined to be the sequence c(P)=(c(v0),m1,c(v1),…,m k ,c(v k ))∈Σk,d.
Given a multi-graph G and a sequence t∈Σk,d for some k, let o c c(t,G) denote the number of paths P in G such that c(P)=t. For an integer , the feature vector f K (G) of level K in G is defined to be the |Σ≤K,d|-dimensional vector f K (G) whose value at each entry t∈Σ≤K,d is given by f K (G)[ t]=o c c(t,G). In this paper, we treat hydrogen-suppressed chemical graphs with carbon C, nitrogen N or oxygen O, which are represented by Σ-colored multi-graphs G with color set Σ={O, N, C }. Figure 2 illustrates an example of Σ-colored multi-graph G that represents a hydrogen-suppressed chemical graph and its feature vector f1(G).
Note that in hydrogen-suppressed chemical graph G, deg(v;G)<val(c(v)) may hold for some vertex v∈V(G). Let us define the residue degree res(v) of a vertex v to be val(c(v))−deg(v;G). In a multi-graph G, we interpret res(v) of a vertex v as the number of hydrogen atoms attached to the vertex v (in our proposed procedure, we also interpret res(v) as the number of new edges/bonds that can be attached to v when G is being constructed by adding more edges).
For a vector of level K≥1, a multi-graph G with f K (G)=g is a multi-graph such that the occurrence of each path t=(c0,m1,c1,…,m p ,c p ) in G with length of at most K is completely specified by g[ t], in particular V(G)={t∣g[ t]≥1,t∈Σ0,d=Σ} (i.e., G has exactly g[ t] vertices of color t), E(G)={t∣g[ t]≥1,t=(c,m,c′)∈Σ1,d} (i.e., G has exactly g[ (c,m,c′)] edges of multiplicity m that join a vertex of color c and a vertex of color c′).
For two vectors and an integer k≥0, let denote the set of all Σ-colored k-augmented trees G such that g L ≤f K (G)≤g U (i.e., f K (G)=g′ for some g with g L ≤g′≤g U ) and deg(v;G)≤val(c(v)), v∈V(G).
Our problem is to enumerate all k-augmented trees G on a given set of atoms each of which is consistent with one of the feature vectors between the lower and upper vectors , such that g L ≤g U (where g L [ t]=g U [ t] for all t∈Σ0,d since the vertex set is fixed for all G).
In what follows, we fix a color set Σ and an upper bound d on multiplicity. We define the problem of enumerating k-augmented trees as follows.
Enumerating chemical graphs with given upper and lower path frequency (EULF)
Given a maximum path length and feature vectors such that g L [ t]=g U [ t] for all t∈Σ0,d, enumerate all multi-graphs .
Figure 3 illustrates an example of an input of EULF with upper and lower feature vectors g L and g U and part of its output, multi-trees and a 1-augmented tree .
For k=0, we have developed an efficient algorithm for EULF [13, 14]. The purpose of this work is to describe an algorithm for EULF with k=1. We assume that the maximum valence is 4 and mainly enumerate a 1-augmented tree such that the cycle contains an edge of multiplicity one (a single bond), since otherwise a 1-augmented tree is a single cycle consisting of edges of multiplicity two, which can be separately handled as a special rare case.
Background
As mentioned in Introduction, enumeration of chemical structures has a long history and many studies have been done. In the field of machine learning, a similar problem, which is called the preimage problem, has been studied [17, 18]. In this problem, a desired object is computed as a feature vector in a feature space, and then the feature vector is mapped back to the input space, where this mapped back object is called a preimage. The definition of the feature vectors based on the frequency of labeled paths [19, 20] or small fragments [10, 21] has been widely used. Akutsu and Fukagawa [22] formulated the graph preimage problem as the problem of inferring graphs from the frequency of paths of labeled vertices and proved that the problem is computationally intractable (NP-hard) even for planar graphs with bounded degrees [22]. Nagamochi [23] proved that a graph determined by the frequency of paths with length one can be found in polynomial time if any exists.
The preimage problem has also been studied in the field of chemoinformatics as a part of inverse QSAR/QSPR (quantitative structure-activity relationship/quantitative structure-property relationship) studies. Indeed, the problem is essentially the same as reconstruction and/or enumeration of molecules from their descriptors in inverse QSAR/QSPR [8, 9, 24, 25], where the descriptors correspond to feature vectors in the preimage problem. Wong and Burkowski developed a practical preimage based method and demonstrated that it actually generated the structure of a new drug candidate [26]. For enumeration of molecules from descriptors, useful tools such as MOLGEN have been developed [27]. However, they are not very efficient if large structures are to be enumerated because many of them treat general graph structures (under the valence constraint).
It might be possible to develop significantly faster algorithms for the preimage problem if we restrict the class of target chemical structures and employ recent techniques for enumeration of graph structures. Fujiwara et al.[28] studied enumeration of tree-like chemical graphs that satisfy a given feature vector which specifies frequency of paths of up to a prescribed length K in a chemical compound to be constructed. They proposed a branch-and-bound algorithm that consists of a branching procedure based on the tree enumeration algorithm by Nakano and Uno [29, 30] and bounding operations designed by properties on path frequency and atom-atom bonds. They showed by means of computational experiments on enumeration of alkane isomers that their algorithm works at least as efficiently as the fastest algorithm while using much less memory space.
To reduce the size of the search space, Ishida et al. [31] have introduced a new bounding operation, called the detachment-cut, based on the result of Nagamochi [23]. In this problem formulation, it is required that the path frequency of a chemical structure is exactly the same as the specified one. However, there does not exist such a structure in many cases because a mapping between chemical structures and feature vectors is not surjective and thus there are many vectors in a feature space that do not have preimages. To seek solutions effectively in a relaxed constraint, Shimizu et al. [13] recently introduced a problem of enumerating tree-like hydrogen-suppressed chemical graphs that satisfy one of a given set of feature vectors which is specified by a pair of upper and lower feature vectors. They proposed a branch-and-bound algorithm for the problem, called 1-Phase algorithm, and afterward Suzuki et al. [14] proposed a more efficient and effective algorithm, called 2-Phase algorithm. Implementations of these algorithms [13, 14] for enumerating tree-like hydrogen-suppressed/hydrogen-retained chemical graphs with given upper and lower bounds on path frequencies are available on a web server ( http://sunflower.kuicr.kyoto-u.ac.jp/tools/enumol2/).
As shown by Nakano and Uno [29, 30], the class of trees admits a nice scheme for computer representation of their structures (called “left-heavy trees”) which enables us to generate trees significantly faster (in constant time per tree) without executing any explicit test on the uniqueness of structure representations of temporarily generated labeled graphs. Development of algorithms for enumerating chemical graphs with a “non-tree structure” is thereby a challenging task if we still wish to attain high computational efficiency as we have achieved for enumeration of tree-like chemical graphs, because no such effective representation scheme is known for general graphs. It should be noted that although polynomial-time algorithms have been developed for equivalence test and unique representation form problems for bounded degree graphs [32, 33] and chemical compounds [34], they are not directly applicable to efficient enumeration of chemical graphs.
In the NCI database ( http://cactus.nci.nih.gov/ncidb2.2/), the ratio of the number of chemical compounds with k-augmented tree structures to that of all registered chemical compounds is approximately 9%, 22%, 28%, 20%, and 11% for k=0,1,2,3, and 4, respectively. This implies that we have been able to treat only 9% of all of chemical compounds with high computational efficiency. As the first step toward efficient enumeration of non-tree chemical graphs, we consider the problem of hydrogen-suppressed chemical graphs with 1-augmented tree (monocyclic) structure. If we can solve this problem, we can treat 31% (=9%+22%) of chemical compounds. Although no effective representation scheme is known even to 1-augmented trees, we can create a tree by removing one edge in the unique cycle in a 1-augmented tree (two multiple edges with the same endvertices is not called a cycle in this paper). Additionally, 2-Phase algorithm [14], which enumerates tree-like hydrogen-suppressed chemical graphs, can be used without any major modification to enumerate such trees T=G−e with one edge deficit from 1-augmented trees G to be constructed. Thus the main task is to efficiently test the uniqueness of generated labeled 1-augmented trees. To design such a procedure, we use a well-reflected definition of a parent 0-augmented tree T=G−e of a 1-augmented tree G. As a result, we can combine the new procedure with 2-Phase algorithm to obtain an algorithm for enumerating hydrogen-suppressed chemical graphs with 1-augmented tree structure from upper and lower bounds on feature vectors.
Method
Our proposed algorithm is based on existing algorithms to enumerate colored trees [29, 30] and colored multi-trees [13, 14, 28, 31]. The basic strategy of our algorithm is to generate a multi-tree first and then extend it to a 1-augmented tree by adding an edge. In enumeration algorithms, it is important not to miss any possible structures and not to duplicate identical structures. In order to efficiently cope with these conditions, the concept of the family tree has been widely employed in various enumeration algorithms. To define a family tree for graphs, we need to define a parent-child relationship between graph structures so that a parent structure is uniquely determined from a child structure, where each child structure is obtained by adding a vertex or an edge to its parent structure. Because extension of a multi-tree to a 1-augmented tree is the core part of our proposed algorithm, we need to provide a proper definition of the parent-child relationship between a multi-tree and a 1-augmented tree. As will be shown later, there may exist multiple possible ways of having a parent structure. How to define the unique parent of a given 1-augmented tree is one of the novel points of our proposed algorithm. Another important issue on generating 1-augmented trees is not to generate identical 1-augmented trees from the same multi-tree. As will be shown later, there is a case in which additions of different edges result in identical structures. How to efficiently prevent this kind of duplicate generation of identical structures is the other novel point of our proposed algorithm. In the following, we give a detailed description of the algorithm including these novel points. Again, readers not interested in mathematical details can skip this part.
Overview of a new algorithm for 1-Augmented trees
Let be the set of 0-augmented trees (multi-trees) T=G−e obtained from each 1-augmented tree by removing a simple edge e in the unique cycle of G.
Then we have for a modified lower vector in a vector set . We construct such a vector set from g L as follows: For each t∈Σk,d with k≥2, let ; and for each t∈Σ1,d, let
Thus our first task is to generate all multi-trees by using fast conventional algorithms such as 2-Phase algorithm.
Our next task is to generate 1-augmented trees G from each multi-tree such that no 1-augmented tree in will be duplicated during the entire enumeration over all . To attain this objective without storing all generated 1-augmented trees for a comparison with a newly generated 1-augmented tree, we define a mapping ; the multi-tree T=π(G) for a 1-augmented tree G is called the parent of G. For a multi-tree T, a 1-augmented tree G with π(G)=T is called a child of T (possibly T has more than one child), and is called a feasible child of T if . Note that any of definition of such a mapping will suffice as long as π(G) is determined only by the information of an “unlabeled graph” G (i.e., topological structure) except for a possible difference in computational efficiency to avoid duplication of solutions.
In the following, we show the 2-Phase algorithm, present details of our definition of parents π and design an efficient procedure for generating all children G from a given multi-tree .
Summary of 2-Phase algorithm for 0-Augmented tree
In this section, we summarize 2-Phase Algorithm [14] for generating all multi-trees in . In the first phase, we simplify input feature vectors by adding the frequencies of the paths that include multiple edges to the corresponding paths which consist of only simple edges and then enumerate simple trees for the simplified upper and lower feature vectors. Figure 5 illustrates feature vectors g U and and simplified feature vectors g u and .

Illustration of modified feature vectors. The frequency of O1C and C1O increases after the simplification.
In the second phase, we assign multiplicities of edges for each of the simple trees to satisfy the feature vector constraint and the valence constraint. The inputs and outputs of the first phase and second phase in 2-Phase Algorithm are described as follows:

From the given multi-tree , our efficient procedure generates all 1-augmented trees in .
Parent-child relationship
In this section, to avoid duplication of a 1-augmented tree during the entire enumeration over all , we introduce a parent-child relationship between a 0-augmented tree and a 1-augmented tree.
Signature of rooted multi-trees
To define the parent π(G) of a 1-augmented tree G using only topological structure, we first introduce the concepts of “canonical form” and “signature” for a class of multi-graphs.
We fix the total order of colors in Σ arbitrarily, e.g., O <N <C, and regard each color c∈Σ as a small integer in . We define the lexicographical order among sequences with elements in as follows. A sequence A=(a1,a2,…,a p ) is lexicographically smaller than a sequence B=(b1,b2,…,b q ) (denoted by A<B) if and only if there is an index k such that (i) a i =b i (1≤i≤k); and (ii) ak+1<bk+1(k+1≤ min{p,q}) or k=p<q; otherwise A=B, i.e., p=q and a i =b i (1≤i≤p), or B<A. Let A≤B denote A<B or A=B.
A multi-graph is called labeled if each vertex has a unique name or an index such as v0,v1,…,vn−1, and we usually record a multi-graph as labeled in our computer. Hence, testing isomorphism of two multi-graphs is to find labels for these “unlabeled graphs” such that the two labeled graphs completely match each other including the adjacency between every two vertices. For a class  of multi-graphs, if we have a way of choosing a label for each multi-graph that is unique up to automorphisms of G, then we can test the isomorphism of two graphs directly with their labels. Such a labeling for G is called the canonical form of G. Once such a canonical form is obtained, we can easily encode each multi-graph into a code σ(G) (which is an integer or a sequence of integers/colors), called the signature of G, such that two multi-graphs are isomorphic if and only if σ(G)=σ(G′). Without loss of generality we assume a total order over by introducing, if necessary, a total order over all colors and a lexicographical (total) order over all sequences of integers and colors.
of multi-graphs, if we have a way of choosing a label for each multi-graph that is unique up to automorphisms of G, then we can test the isomorphism of two graphs directly with their labels. Such a labeling for G is called the canonical form of G. Once such a canonical form is obtained, we can easily encode each multi-graph into a code σ(G) (which is an integer or a sequence of integers/colors), called the signature of G, such that two multi-graphs are isomorphic if and only if σ(G)=σ(G′). Without loss of generality we assume a total order over by introducing, if necessary, a total order over all colors and a lexicographical (total) order over all sequences of integers and colors.
A rooted graph is a multi-graph in which a vertex is designated as the root, and two rooted graphs are isomorphic if there is an isomorphism that maps their roots onto each other.
Any tree T has either a vertex or a pair of adjacent vertices removal of which leaves no component with at least |V(T)|/2 vertices [35], where the former is called the centroid and the latter is called the bicentroid.
In a rooted multi-tree T, the parent vertex of a non-root vertex v is denoted by p(v) and the depth of a vertex v is denoted by depth(v), where the depth of a vertex is its distance to the root. For a vertex v in T, let T v denote the subtree induced from T by all descendants of v including v. For an edge e=u v in T (where u=p(v)), let T e (T u v ) denote the subtree of T that consists of T v and u=p(v) joined by edge e=u v.
For the class of rooted multi-trees, a canonical form of a rooted multi-tree T is given by an “ordered tree” τ of it (i.e., determination of a total order among children of each vertex). Let dfs(τ) denote the total order of vertices in τ visited by the depth-first-search order according to the order for children in τ. For example, Figure 6 illustrates three ordered trees τ1, τ2 and τ3, which are obtained from the same multi-tree T rooted at the centroid, where the number beside each vertex v indicates dfs(v).

Illustration of a rooted tree and left-heavy trees. (a) An ordered tree τ1 rooted at its centroid; (b) a left-heavy tree τ2; and (c) the canonical form τ3.
We let δ(τ) denote the alternating sequence (c0,d0,c1,d1,…,cn−1,dn−1) such that c i and d i denote the color and depth, respectively, of the i-th vertex v i in dfs(τ), and M(τ) denote the sequence (m1,m2,…,mn−1) of the multiplicity m i =m(v i ,p(v i )) of the edge joining the i-th vertex and its parent p(v i ) in T. For a vertex v, let dfs(v) denote the labeling number of v in dfs(τ). For example, Figure 6 illustrates δ(τ i ) of ordered trees τ i , i=1,2,3 and M(τ2) and M(τ3).
A left-heavy tree of a rooted multi-tree T is an ordered tree τ that has the maximum code δ(τ) among all ordered trees of T (hence a left-heavy tree τ is a canonical form and δ(τ) is a signature of it when we ignore the multiplicity of rooted multi-trees). We define the canonical form of a rooted multi-tree T to be the left-heavy tree τ that has the maximum code M(τ) among all left-heavy trees of T, and let σ(T) denote a signature of T (a code of the canonical form τ such as (δ(τ),M(τ))). For example, in Figure 6, τ2 and τ3 are left-heavy trees of T, since they have lexicographically maximum sequences δ(τ2)=δ(τ3) among all ordered trees τ of the rooted multi-tree T, and τ3 is the canonical form of T and (δ(τ3)=(C,0,C,1,N,2,C,1,N,2,N,1,O,2),M(τ3)=(2,1,1,2,1,1)) is the signature of T since it is a left-heavy tree with the lexicographically maximum M(τ3) among all left-heavy trees τ of T.
Using the canonical form for rooted multi-trees, we can define a canonical form for “unrooted” multi-trees T by regarding them as trees rooted at the centroid or bicentroid.
Defining parents π
We are now ready to define parents π for 1-augmented trees (note that there is no root for any 1-augmented tree). Let G be a 1-augmented tree with a unique cycle C of length p which by our assumption contains at least one simple edge. Then there are p possible choices G−e i , i=1,2,…,p, for the parent of G. We introduce a rule to choose one of them based only on the topological information on G and C. For each vertex v in C, let N(v) denote the set of vertices in V−V(C) adjacent to v. Removing an edge vw with v∈V(C) and w∈N(v) leaves a multi-tree containing w, which we denote by T w . For each vertex v in C, we encode all multi-trees T w , w∈N(v) into a signature σ∗(v) using the signature σ for rooted multi-trees; we set
such that holds for N(v)={w1,w2,…,w h }. Note that two vertices v and v′ in C have the same color and an identical set of subtrees in N(v) and N(v′) if and only if σ∗(v)=σ∗(v′). For each simple edge e=u v in C, we define a code c∗(e) as follows. We encode the unique path u1 (=u),u2,…,u h (=v) from u to v along C into σ∗(u,v)=(σ∗(u1),m1,σ∗(u2),m2,…,mh−1,σ∗(u h )), where m i =m(u i ,ui+1). Symmetrically, we define σ∗(v,u) = (σ∗(u h ),mh−1,σ∗(uh−1),mh−2,…,m1,σ∗(u1)). The code c∗(e) is defined to be lexicographically the maximum one between two sequences σ∗(u,v) and σ∗(v,u). Furthermore, let E∗(C) be the set of simple edges e∗ in C such that c∗(e∗) is lexicographically maximum among c∗(e) for all simple edges e in C.
We call an edge vw with v∈V(C) and w∈N(v) a heavy edge if T w has at least |V(G)|/2 vertices. We distinguish two cases to define parent π.
-
Case 1.
There is no heavy edge around C: For an arbitrary edge e∈E∗(C), we define π(G) to be G−e (note that when |E∗(C)|≥2, G is symmetric around C and G−e and G−e′ will be isomorphic for any two edges e,e′∈E∗(C)). Figure 7 illustrates how the parent π(G) of a 1-augmented tree G in Case 1 is determined on these signatures σ(u,v) and σ(v,u) of simple edges uv in the cycle C.
Figure 7 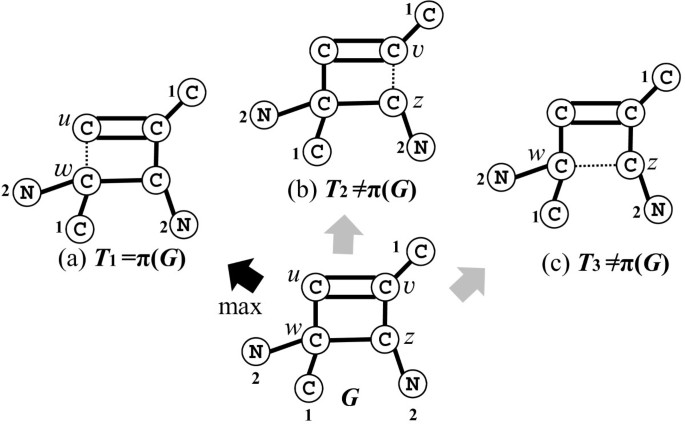
Illustration of defining the parent π ( G ) of a 1-augmented tree G . Three multi-trees T1=G−u w, T2=G−v z and T3=G−w z are obtained from G by removing a simple edge in the cycle. Each number on the left side of each vertex v in G indicates its signature σ∗(v) of {T w ∣w∈N(v)}. The code σ∗ for each pair of adjacent vertices in the cycle of G is given by σ∗(w,u)=((C,2,1),1,(C,2),1,(C,1),2,(C)), σ∗(u,w)=((C),2,(C,1),1,(C,2),1,(C,2,1)), σ∗(v,z)=((C,1),2,(C),1,(C,2,1),1,(C,2)), σ∗(z,v)=((C,2),1,(C,2,1),1,(C),2,(C,1)), σ∗(w,z)=((C,2,1),1,(C),2,(C,1),1,(C,2)), and σ∗(z,w)=((C,2),1,(C,1),2,(C),1,(C,2,1)). Then π(G) is defined to be T1=G−u w because σ∗(w,u) is maximum over all of these six codes.
-
Case 2.
There is a heavy edge v∗w∗: Note that no other edge can be a heavy edge. Let e1 and e2 be the two edges in C that are adjacent to v∗, where at least one of them is a simple edge since deg(v)≤4. If exactly one of them, e.g., e1 is a simple edge, then we define π(G) to be G−e1. When e1 and e2 are simple edges, we choose one of them as follows. We first ignore all trees T w with w∈N(v∗), which are symmetric at the vertex v∗ commonly shared by e1 and e2 and hence useless to construct a signature for distinguishing e1 and e2. Without using T w with w∈N(v∗), we construct the code c∗(e1) and c∗(e2). Finally we choose any edge e i such that c∗(e i ) is lexicographically maximum between c∗(e1) and c∗(e2), and define π(G) to be G−e i .

Generating children
Recall that our algorithm for enumerating 1-augmented trees consists of two major stages: the first stage enumerates all multi-trees by 2-Phase algorithm, and the second stage generates all feasible children G for each , i.e., 1-augmented trees with π(G)=T. This section describes a procedure for generating all children G=T+e of a given multi-tree T by adding a new edge e.
For simplicity, we consider the case where a given multi-tree T has the centroid (the case where it is rooted at the bicentroid can be treated with a minor technical modification). In the following, we assume that a given multi-tree T is represented as its canonical form (a left-heavy tree) τ rooted at its centroid, and that its sequences δ(τ)=(c1,d1,…,c n ,d n ) and M(τ)=(m2,m3,…,m n ) over the labeling dfs(τ) have been already computed after the first stage (2-Phase algorithm can deliver not only solutions T but also τ and these sequences together).
It should be noted that the canonical form of left-heavy trees enjoys the following recursive structure. For any vertex v in T, the subtree T v of T rooted at v induces an ordered tree τ v from the left-heavy tree τ and τ v is again the canonical form of T v , since dfs(τ v ) is a subsequence of dfs(τ) with consecutive vertices and its ordered pair (δ(τ v ),M(τ v )) is also lexicographically maximized over all ordered trees of T v .
Testing generated 1-Augmented trees
Given the left-heavy tree τ of a multi-tree T, we add a new edge xy for two nonadjacent vertices x,y∈V(T)(dfs(x)<dfs(y)) to obtain G=T+x y. Let C denote the cycle created in G. We check the following condition to test whether T is the parent of Gor not.
-
Case I.
C contains the root (centroid) of T:
-
(A)
σ∗(x y) is lexicographically maximum among σ∗(e) for all simple edges e in C.
-
(A)
-
Case II.
Otherwise:
-
(B)
x is the ancestor of y;
-
(C)
σ∗(x y)≥σ∗(e) if the edge e incident to x in C is simple.
-
(B)
Then, we have the following lemma, where the proof is given in S1.1 (of the Additional file 1).
Lemma 1.
For a multi-tree T and two nonadjacent vertices x,y∈V(T), testing whether T=π(T+x y) can be done by checking the above condition in O(|V(C)|2) time.
Avoiding duplication of children
In previous sections, we have showed that all children (i.e., 1-augmented trees) of a given multi-tree T can be generated by the definition of the parent-child relationship between multi-tree and 1-augmented tree. However, if T has two isomorphic subtrees T u and T v then we would have two children T+x y and T+x′y′, which are isomorphic to each other. To avoid such duplication, we test if T+x y is isomorphic to T+x′y′ for some other vertices x′ and y′ when we add an edge xy to T. In fact, we do not try to find other such pair x′ and y′ explicitly. Instead we introduce a rule that we do not generate T+x y by any edge xy that has such an “isomorphic” vertex pair x′ and y′ on the left hand side of x and y in T. To detect this situation efficiently, we first compute data on each vertex v in T that indicates whether the left hand side of v contains another vertex u such that its subtree Tp(u)u is isomorphic to Tp(v)v. Using such data, we show that given a vertex pair x and y whether there is an isomorphic pair x′ and y′ in the left hand side can be checked in constant time. In other words, we show an O(n2) time algorithm extracting all the leftmost side vertex pairs from T.Among all isomorphic vertex pairs, we call the leftmost one an “admissible pair”, where “isomorphic” means here that the connection of vertices in each pair results in an isomorphic tree. Figure 8 shows an example of a case in which additions of different edges result in identical structures and the admissible pair.

Illustration of admissible and non-admissible pairs. A cycle will be created by adding each edge shown by a black dotted line. Two 1-augmented trees T+u v and T+u v′ are 1-augmented trees obtained from the multi-tree T by adding a simple edge such that T+u v and T+u v′ are isomorphic to each other. (a) The vertex pair (u,v) is an admissible pair, and (b) the other is not an admissible pair (at least one admissible pair always exists in every 1-augmented tree). Therefore, T+u v′ is not created (all 1-augmented trees are discarded except for the 1-augmented tree created for admissible pair).
In this section, we show the validity that we only need to add an edge between each admissible pair to avoid duplication and omission of 1-augmented trees generated from one multi-tree T. Finally, for a multi-tree, we provide an efficient algorithm extracting all vertex pairs to generate all children of the multi-tree.
Admissible pairs
We write T+u v∼T+u′v′ if and only if T+u v and T+u′v′ are isomorphic. For a tree T, let c T denote its centroid, which is either a vertex (unicentroid) or an edge (bicentroid). Let T be a left-heavy tree rooted at its centroid c T . When c T is a bicentroid r r′, r and r′ will be the vertices that have no parent in the parent-child relationship in T. We shall now introduce “rooted-isomorphism” among 1-augmented trees obtained from T by adding a new edge. We regard a 1-augmented tree G=T+u v obtained by adding new edge uv between two nonadjacent vertices u,v∈V(T) as a graph rooted at c T . When c T is a vertex r, we say that two 1-augmented trees G=T+u v and G′=T+u′v′ are rooted-isomorphic if they admit an isomorphism ψ such that c T in G=T+u v corresponds to c T in G′=T+u′v′ (i.e., ψ(r)=r when c T is a vertex r, and {ψ(r),ψ(r′)}={r,r′} when c T is an edge r r′). We write if and only if T+u v and T+u′v′ are rooted-isomorphic with root r. Then, the following theorem holds, where the proof is given in Additional file 1: S1.2.
Theorem 2.
Let T be a left-heavy tree rooted at its centroid c T and {u,v},{u′,v′}⊆V(T) be two pairs of nonadjacent vertices. If T+u v∼T+u′v′ then .
Theorem 2 tells us that two 1-augmented trees G=T+u v and G′=T+u′v′ are isomorphic if and only if they are rooted-isomorphic (i.e., c T in G corresponds to c T in G′ in the isomorphism ψ, where possibly ψ(r)=r′ and ψ(r′)=r when c T =r r′).
Now we consider how to generate a set of 1-augmented trees T+u v such that the 1-augmented tree T+u v for any pair of nonadjacent vertices u,v∈V(T) is isomorphic to exactly one 1-augmented tree G in the set . By Theorem 2, we only need to check the rooted-isomorphism among 1-augmented trees T+u v for all pairs of nonadjacent vertices u,v∈V(T). Based on this, we can modify a given tree T with bicentroid c T =r r′ into a tree T′ with unicentroid r∗ by inserting a new vertex on the edge r r′. Since this does not change the rooted-isomorphism among 1-augmented trees T′+u v or the left-heaviness of T, we assume in the following that a given tree T has a unicentroid c T =r.
Let T be a left-heavy tree. We shall introduce some terminology. Let x be a non-root vertex x in T. Denote by left(x) the immediate left sibling of a non-root vertex x (if any). We define data copy as follows.
Let u and v be two vertices in T. We denote by P(u,v) the unique path in T that connects u and v, where P(u,v)=P(v,u). Let lca(u,v) denote the least common ancestor of u and v, i.e., the highest vertex in P(u,v) (where we define lca(u,v) to be the edge c T =r r′ when T is rooted at the bicentroid c T =r r′, and u∈V(T r ) and ). When dfs(u)<dfs(v), we define the greatest uncommon ancestor gua of u and vas follows:
● Let gua(u,v) denote the child of lca(u,v) that is closest to u in T, where gua(u,v) is an ancestor of u (including u itself) if lca(u,v)≠u;
● Let gua(v,u) denote the child of lca(u,v) that is an ancestor of v (including v itself), where gua(u,v)=gua(v,u) if lca(u,v)=u.
We call a pair of nonadjacent vertices u,v∈V(T) with dfs(u)<dfs(v)admissible if it satisfies the following conditions (see Figure 9 for conditions (1) and (2) and Figure 10for condition (3)):
-
(1)
copy(w) = 0 for all vertices w∈ V(P(lca(u,v),r))−{r};
-
(2)
copy(w)=0 for all vertices w∈V(P(u,gua(u,v)))∪V(P(v,gua(v,u)))−{lca(u,v),gua(v,u)};
-
(3)
if copy(gua(v,u))=1 then
-
(i)
gua(u,v)=left(gua(v,u)) (hence u≠lca(u,v)); and
-
(ii)
For the copy of vertex u in Tgua(v,u), it holds (where ).
-
(i)

Illustration of conditions (1) and (2) for admissible pairs. The black dotted line joins two vertices u and v, which will be the edge to create a cycle in the 1-augmented tree T+u v. (a) and (b) illustrate the case of lca(u,v)≠u (where two rooted subtrees T(gua(u,v)) and T(gua(v,u)) are not isomorphic to each other by copy(gua(v,u))=0) and the case of lca(u,v)=u, respectively (note that lca(u,v)≠v by dfs(u)<dfs(v)). Conditions (1) and (2) exclude any ancestor w of u or v such that copy(w)=1 (otherwise we can prove that there is a lexicographically smaller pair (u′,v′) such that T+u′v′ is isomorphic to T+u v).

Illustration of condition (3) for admissible pairs. The black dotted line joins two vertices u and v, which will be the edge to create a cycle in the 1-augmented tree T+u v. Condition (1) and (2) exclude any ancestor w of u or v such that copy(w)=1 except for w=gua(v,u). The assumption copy(gua(v,u))=1 requires the rooted subtree Tgua(u,v) to be isomorphic to Tgua(v,u) and condition (3)-(i) requires Tgua(u,v) to be located immediately on the left of Tgua(v,u) among the subtrees rooted at the children of lca(u,v). Then Tgua(v,u) contains a copy of u (i.e., ). Similarly Tgua(u,v) contains a copy of v (i.e., ). Condition (3)-(ii) requires (otherwise we can prove that is a lexicographically smaller pair such that is isomorphic to T+u v). Although Tgua(u,v) and Tgua(v,u) are isomorphic to each other, only paths and nodes relevant for explanation are shown in this figure.
Note that (3)-(i) implies that copy(gua(v,u)) in (2) needs to be 0 when lca(u,v)=u.
The next lemma indicates that we only need to add an edge between each admissible pair to avoid duplication of 1-augmented trees, where the proof is given in Additional file 1: S1.3.
Lemma 3.
For a left-heavy tree T rooted at its unicentroid c T =r, let admissible pairs u,v∈V(T)}. Then the 1-augmented tree T+u v for any pair of nonadjacent vertices u,v∈V(T) is isomorphic to exactly one 1-augmented tree G in .
Algorithm
In this section, we describe an algorithm of the second stage to generate all children of a given multi-tree T without duplication and omission, and show the computational complexity of the second stage. To generate all children of T, we first find all admissible pairs (u,v) for T and test whether T is the parent π(T+u v) of T+u v or not. Notice that a straightforward method would take O(n) time to check whether a pair (u,v) is admissible or not. Since there are at most n C2 vertex pairs in a multi-tree T, finding all admissible pairs for T may take O(n3) time. That is, from Lemma 1, we may need O(n3|V(C)|2)=O(n5) time to generate all children of T.
In what follows, we design a faster O(n4)-time algorithm to generate all children of a given multi-tree T. For this, we find only a subset of all admissible pairs, called the set of “candidate” pairs defined as follows (see also Figure 11). We see that no pair (x,y) generates a child T+x y of T if a heavy edge is created in T+x y and x is not an ancestor of y, since such (x,y) does not satisfy any of Cases I and II for generating children of T. Hence we are not interested in storing such pairs (x,y), and call an admissible pair (x,y) a candidate pair when (i) no heavy edge is created in T+x y; or (ii) x is an ancestor of y, where (i) (resp., (ii)) is a necessary condition of Case I (resp., Case II). By definition, every candidate vertex pair (x,y) is admissible, whereas any admissible pair (x,y) such that T+x y is a child of T is always a candidate pair. Therefore, to generate all children of T, we do not need to find all admissible pairs and only have to extract all candidate pairs.

Illustration of candidate pairs. A graph G will be created by adding each edge shown by a black dotted line. However, no vertex pair for any of edges a,b and c is a candidate pair. We do not have to add edges a,b, and c to T1,T2, and T3, respectively because neither G−a, G−b, nor G−c can be the parent of G.
To facilitate this, we examine all vertex pairs (u,v) (dfs(u)<dfs(v)) in T in a lexicographical order with respect to (dfs(u),dfs(v)), i.e., we choose each vertex v i from v0 to vn−1 as u and then choose each vertex v j from vi+1 to vn−1 as v. We call the lexicographical order over vertex pairs a dfs order. For each of the generated vertex pairs, we check whether it is a candidate pair or not. Finally, for each candidate pair (u,v), we test whether T+u v is a child of T in Case I or II.
We can find all candidate pairs for a multi-tree T in O(n2) time in total as stated below. The proof is given in Additional file 1: S1.4.
Lemma 4.
For a left-heavy multi-tree T, all candidate pairs of T can be found in O(n2) time.
Finally, by Lemma 1 and Lemma 4, we can generate all children of a multi-tree T in O(n4) time, as stated in the next lemma.
Lemma 5.
Given a left-heavy multi-tree T, all children of T can be generated in O(n4) time.
Proof.
For a left-heavy multi-tree T, we can find all candidate pairs in O(n2) time by Lemma 4. For each candidate pair (u,v), we can test whether T=π(T+u v) or not in O(|V(C)|2) time by Lemma 1. Thus, for a left-heavy multi-tree T, all children of T can be generated in O(n2|V(C)|2)=O(n4) time. □
Experimental and results
This section reports experimental results of our algorithm enumerating 1-augmented trees. Tests were carried out on a PC with an Intel Core i5 processor running at 3.20 GHz and the Linux operating system using the C language, employing instances based on chemical compounds selected from the KEGG LIGAND database [15] ( http://www.genome.jp/ligand/).
-
(I)
First we select four chemical compounds “C00062,” “C03343,” “C03690,” and “C07178” as chemical graphs with 0-augmented tree (acyclic) structure and four chemical compounds “C00095,” “C00270,” “C00645,” and “C00837” as chemical graphs with 1-augmented tree structure (see Additional file 1: Figure S21 for illustrations of these chemical graphs), wherein each benzene ring in chemical compounds “C03343,” “C03690,” and “C07178” is regarded as a virtual atom b of valence 6. These compounds are heuristically selected based on the following criteria: (i) each compound is a 0-augmented tree or 1-augmented tree (except benzene ri ngs), (ii) each compound consists of C,O,H (or, C,O,N,H) atoms, (iii) compounds are not very similar to each other, and (iv) compounds have varying sizes but are not too large.
The virtual atom b is treated as one atom so that we discard all possible regioisomers of benzene. Thus in our experiment, we consider the cycles not caused by benzenes but by other substructures in these 1-augmented trees. We remark that an efficient algorithm has been developed for generating all possible regioisomers of a given 0-augmented tree structure with virtual atoms b by Li et al. [36], and an implementation of the algorithm is available on a web server ( http://sunflower.kuicr.kyoto-u.ac.jp/tools/enumol2/).
To generate problem instances from each of the selected chemical graphs, we define to be a width between upper and lower feature vectors. From the feature vector g=f K (G) of a chemical graph G at level K, we construct two feature vectors g U and g L of width w as follows. For each entry t with g[ t]≥1, let g U [ t]=g[ t]+w and g L [ t]= max{g[ t]−w,0}; and for each entry t with g[ t]=0, let g U [ t]=g L [ t]=0. See Additional file 1: Figure S23 (resp., Additional file 1: Figure S24) for the lower and upper feature vectors g L and g U with K=1 and w=1 (resp., K=2 and w=1) created from C00062.
To examine the computational efficiency, we compare the time per output multi-tree/1-augmented tree by our algorithm and by 2-Phase algorithm [14]. Our algorithm enumerates not only the 1-augmented trees in but also the multi-trees in . Therefore, if time per output graph of our algorithm is close to that of 2-Phase algorithm, then we can enumerate 1-augmented trees as fast as 2-Phase algorithm enumerates multi-trees.
Table 1 shows the result of the comparison of 2-Phase algorithm and our algorithm for varying K with fixed w=1, where the meanings of columns are as follows.
Note on tables:
-
(1)
C00062, C00095, C00270 C00645, C00837, C03343, C07178, and C03690 are the chemical compounds in the KEGG LIGAND database, respectively;
-
(2)
in Table 1, the width for constructing upper and lower feature vectors is 1;
-
(3)
n is the number of vertices without hydrogen atoms in an instance preprocessed by replacing each benzene ring with a new atom with six valences;
-
(4)
w is the width for constructing upper and lower feature vectors;
-
(5)
K is the level of given feature vectors;
-
(6)
“time (s)” is the CPU time in seconds;
-
(7)
T.O. means the “time over” (the time limit is set to be 1,800 seconds);
-
(8)
“time/graph” is the time per enumerating one graph;
-
(9)
“tree” is the number of all possible solutions of tree-like chemical graph in the time limit;
-
(10)
“cycle” is the number of all possible solutions of 1-tree chemical graph in the time limit;
-
(11)
“ratio” is a number such that “time/graph” of our algorithm is divided by that of 2-Phase algorithm;
-
(12)
for any real numbers x and y, let xEy denote x×10 y.
It is to be noted that in some instances, the number of enumerated trees by 2-Phase algorithm and that of our algorithm are different because of the time limit. Hence, the “tree” and “cycle” columns show the number of incomplete solutions in instances whose “time” column is “T.O.”. However, this is not a critical issue because we mainly want to know the “time per graph” and its “ratio” between 2-Phase algorithm and our algorithm. We can make use of them as beneficial results from “tree,” “cycle,” and “time” columns even if they are incomplete and “T.O.”.
We find that almost all instances solved within the time limit by 2-Phase algorithm are also solved by our algorithm within the time limit. Moreover, the “ratio” of instances is less than 10 except 4 out of 38 cases, and that of many instances is less than 5. This means that the time per output by our algorithm is close to that by 2-Phase algorithm. Therefore, we have demonstrated that our algorithm maintains the high computational efficiency of 2-Phase algorithm even if K changes. Note that our algorithm does not output any 1-augmented trees in in “C00062,” “C03343,” “C07178,” and “C03690” when K is large. This is because the instances are acyclic chemical compounds: 1-augmented trees become less able to satisfy the feature vector constraint as K increases and only multi-trees can satisfy the feature vector constraint.
Table 2 shows the result of the comparison of 2-Phase algorithm and our algorithm for varying w with fixed K=3. Just like with Table 1, almost all instances solved within the time limit by 2-Phase algorithm are also solved by our algorithm within the time limit. The “ratio” of instances is less than 10 except 7 out of 48 cases, and that of many instances is less than 5. In particular, with respect to “C00095,” “C00645,” and “C00837,” which have 1-augmented tree structure, the “ratio” is less than 1 or close to 1. This implies that our algorithm can enumerate 1-augmented trees and multi-trees faster than 2-Phase algorithm enumerates multi-trees. These results mean that the time per output by our algorithm is close to that by 2-Phase algorithm. Therefore, we have demonstrated that our algorithm maintains the high computational efficiency of 2-Phase algorithm even if w changes.
Finally, from Table 1 and Table 2, we compare 2-Phase algorithm and our algorithm in terms of varying n, where n is the size of an instance. Note that n is the number of vertices without hydrogen atoms in an instance preprocessed by replacing each benzene ring with a new atom with six valences. We notice that there is no large difference in the “ratio” between all cases in spite of the fact that the instance size of C03690 is almost twice as large as that of C00062. This implies that our algorithm maintains the high computational efficiency of 2-Phase algorithm even if the instance size becomes large.
-
(II)
Next we select four chemical compounds, prostaglandin (D08040), allobarbital (D00332), gabapentin (D00555), and histamine (D00079) as chemical graphs with 1-augmented tree structure (see Additional file 1: Figure S22 for illustrations of these chemical graphs), all of which are existing drug compounds. We conducted the same experiment as we did for (I): Table 3 shows the result of the comparison of 2-Phase algorithm and our algorithm for varying K with fixed w=1; Table 4 shows the result of the comparison of 2-Phase algorithm and our algorithm for varying w with fixed K=3. In this experiment, we observe that there still is no large difference in the “ratio” between all cases except for the instance of D00079.
Discussions and conclusions
We considered the problem of enumerating all chemical graphs of 1-augmented tree structure from a given set of path-frequency based feature vectors specified by upper and lower feature vectors, and proposed a new exact algorithm by extending 2-Phase algorithm [14]. The experimental results reveal that the computational efficiency of the new algorithm remains high, considering the hardness of treating 1-augmented trees compared with 0-augmented trees.
One of our future works is to introduce new bounding operations for 1-augmented trees in 2-Phase algorithm and our procedure for creating a cycle. Additionally, it is important to extend the proposed algorithm for enumeration of k-augmented trees with k≥2 because we can cover 59%, 79%, and 90% of chemical compounds by 2-augmented trees, 3-augmented trees, and 4-augmented trees, respectively. In this paper, we used the assumption that chemical graphs we treat contain only atoms with valence at most 4 (except benzene rings) in order to define the parent of a 1-augmented tree G as a 0-augmented tree T that is obtained by removing an edge corresponding to a single bond in G. However, it is not difficult to extend our enumeration algorithm for chemical graphs possibly with atoms with valence more than 4 just by modifying the definition so that the parent of a 1-augmented tree G is allowed to be a 0-augmented tree T obtained by removing an edge that corresponds to a double or triple bond in G. Although benzene rings have already been treated as virtual atoms of valence 6, regioisomers are ignored in the proposed algorithm. As mentioned in “Experimental and results” section, an efficient algorithm for generating all possible regioisomers of a given 0-augmented tree structure with virtual atoms b has been developed [36]. Therefore, combination of the proposed algorithm with that algorithm is left as future work as well as further extensions for including atoms with valence more than 4 and furan and more general structures.
Although we do not aim to develop enumeration algorithms that are directly applicable to drug design, this is a future target of our research. In order to apply enumeration algorithms to drug design, considering features based on the path frequency is far from sufficient. Factors such as hydrogen bond donors, hydrogen bond acceptors, positive charges, negative charges, and hydrophobic centers should be taken into account. In addition, the binding site information of the target molecule and geometric information such as the occurrence of rotatable bonds should be reflected. In order to include these factors in enumeration algorithms, we should develop efficient methods that can relate chemical graphs with such physico-chemical and geometric factors. However, such a development is not an easy task even for one type of factor and thus is long-term future work.
References
Bytautas L, Klein DJ: Chemical combinatorics for alkane-isomer enumeration and more. J Chem Inf Comput Sci. 1998, 38: 1063-1078. 10.1021/ci980095c.
Bytautas L, Klein DJ: Formula periodic table for acyclic hydrocarbon isomer classescombinatorially averaged graph invariants. Phys Chem Chem Phys. 1999, 1: 5565-5572. 10.1039/a906137a.
Bytautas L, Klein DJ: Isomer combinatorics for acyclic conjugated polyenes: enumeration and beyond. Theoret Chem Acc. 1999, 101: 371-387. 10.1007/s002140050455.
Buchanan BG, Feigenbaum EA: DENDRAL and Meta-DENDRAL: their applications dimension. Artif Intell. 1978, 11: 5-24. 10.1016/0004-3702(78)90010-3.
Funatsu K, Sasaki S: Recent advances in the automated structure elucidation system, CHEMICS. Utilization of two-dimensional NMR spectral information and development of peripheral functions for examination of candidates. J Chem Inf Comput Sci. 1996, 36: 190-204. 10.1021/ci950152r.
Fink T, Reymond JL: Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery. J Chem Inf Comput Sci. 2007, 47: 342-353. 10.1021/ci600423u.
Mauser H, Stahl M: Chemical fragment spaces for de novo design. J Chem Inf Comput Sci. 2007, 47: 318-324. 10.1021/ci6003652.
Faulon JL, Churchwell CJ: The signature molecular descriptor. 2. Enumerating molecules from their extended valence sequences. J Chem Inf Comput Sci. 2003, 43: 721-734. 10.1021/ci020346o.
Hall LH, Dailey ES: Design of molecules from quantitative structure-activity relationship models. 3. Role of higher order path counts: Path 3. J Chem Inf Comput Sci. 1993, 33: 598-603. 10.1021/ci00014a012.
Deshpande M, Kuramochi M, Wale N, Karypis G: Frequent substructure-based approaches for classifying chemical compounds. IEEE Trans Knowl Data Eng. 2005, 17: 1036-1050.
Cayley A: On the analytic forms called trees with applications to the theory of chemical combinations. Rep Br Assoc Adv Sci. 1875, 45: 257-305.
Pólya G: Kombinatorische anzahlbestimmungen für gruppen, graphen, und chemische verbindungen. Acta Math. 1937, 68: 45-254.
Shimizu M, Nagamochi H, Akutsu T: Enumerating tree-like chemical graphs with given upper and lower bounds on path frequencies. BMC Bioinformatics. 2011, 12 (Suppl 14): 53-
Suzuki M, Nagamochi H, Akutsu T: A 2-phase algorithm for enumerating tree-like chemical graphs satisfying given upper and lower bounds. IPSJ SIG Technical Reports. BIO 2012, 2012, 28 (17): 1-8.
Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M: KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38: D355-D360,
Harary F: Graph Theory. 1969, MA: Addison Wesley
Bakir GH, Weston J, Schölkopf B: Learning to find pre-images. Adv Neural Inform Process Syst. 2003, 16: 449-456.
Bakir GH, Zien A, Tsuda K: Learning to find graph pre-images. Lect Notes Comput Sci. 2004, 3175: 253-261. 10.1007/978-3-540-28649-3_31.
Kashima H, Tsuda K, Inokuchi A: Marginalized kernels between labeled graphs. Proceedings of the 20th International Conference Machine Learning. 2003, Palo Alto: AAAI Press, 321-328.
Ueda N, Akutsu T, Perret JL, Vert JP, Mahé P: Graph kernels for molecular structure-activity relationship analysis with support vector machines. J Chem Inf Model. 2005, 45: 939-951. 10.1021/ci050039t.
Byvatov E, Fechner U, Sadowski J, Schneider G: Comparison of support vector machine and artificial neural network systems for drug/nondrug classification. J Chem Inf Comput Sci. 2003, 3: 1882-1889.
Akutsu T, Fukagawa D, Jansson J, Sadakane K: Inferring a graph from path frequency. Discrete Appl Math. 2012, 160: 1416-1428. 10.1016/j.dam.2012.02.002.
Nagamochi H: A detachment algorithm for inferring a graph from path frequency. Algorithmica. 2009, 53: 207-224. 10.1007/s00453-008-9184-0.
Kier L, Hall L, Frazer J: Design of molecules from quantitative structure-activity relationship models. 1. Information transfer between path and vertex degree counts. J Chem Inf Comput Sci. 1993, 33: 143-147. 10.1021/ci00011a021.
Miyao T, Arakawa M, Funatsu K: Exhaustive structure generation for inverse-QSPR/QSAR. Mol Inform. 2010, 29: 111-125. 10.1002/minf.200900038.
Wong WWL, Burkowski FJ: A constructive approach for discovering new drug leads: Using a kernel methodology for the inverse-QSAR problem. J Cheminf. 2009, 1: 4-10.1186/1758-2946-1-4.
Gugisch R, Kerber A, Laue R, Meringer M, Rucker C: History and progress of the generation of structural formulae in chemistry and its applications. MATCH Comm Math Comput Chem. 2007, 58: 239-280.
Fujiwara H, Wang J, Zhao L, Nagamochi H, Akutsu T: Enumerating tree-like chemical graphs with given path frequency. J Chem Inf Model. 2008, 48: 1345-1357. 10.1021/ci700385a.
Nakano S, Uno T: Generating colored trees. Lect Notes Comput Sci. 2005, 3787: 249-260. 10.1007/11604686_22.
Nakano S, Uno T: Efficient generation of rooted trees. NII Technical Report. 2003, NII-2003-005E,
Ishida Y, Zhao L, Nagamochi H, Akutsu T: Improved algorithms for enumerating tree-like chemical graphs with given path frequency. Genome Inform. 2008, 21: 53-64.
Luks EM: Isomorphism of graphs of bounded valence can be tested in polynomial time. J Comput Syst Sci. 1982, 25: 42-65. 10.1016/0022-0000(82)90009-5.
Fürer M, Schnyder W, Specker E: Normal forms for trivalent graphs and graphs of bounded valence. Proceedings of 15th Annual ACM Symp Theory of Computing. 1983, NY: ACM Press, 161-170.
Faulon J-L: Isomorphism, automorphism partitioning, and canonical labeling can be solved in polynomial-time for molecular graphs. J Chem Inf Comput Sci. 1998, 38: 432-444. 10.1021/ci9702914.
Jordan C: Sur les assemblages de lignes. J Reine Angew Math. 1869, 70: 185-190.
Li J: Enumerating benzene isomers of tree-like chemical graphs. Master’s Thesis. Department of Applied Mathematics and Physics, Graduate School of Informatics, Kyoto University 2014,
Acknowledgements
This work was supported in part by Grant-in-Aid for Scientific Research (KAKENHI) no. 22240009 from JSPS, Japan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HN designed the algorithm and performed theoretical analysis on it. MS detailed the algorithm, implemented it, and performed computational experiments. TA participated in discussions for analyses of the algorithm and the results of computational experiments. HN and MS wrote the manuscript and TA revised it. All authors read and approved the final manuscript.
Electronic supplementary material
13321_2013_606_MOESM1_ESM.pdf
Additional file 1: Proofs of Lemmas and Theorems. Proofs of Lemma 1, Theorem 2, Lemma 3, and Lemma 4, descriptions of the 2-phase algorithm and the main algorithm, and some figures for chemical graphs and upper and lower feature vectors used in the computational experiment are given in this supplemen (PDF 2 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article

Cite this article
Suzuki, M., Nagamochi, H. & Akutsu, T. Efficient enumeration of monocyclic chemical graphs with given path frequencies. J Cheminform 6, 31 (2014). https://doi.org/10.1186/1758-2946-6-31
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1758-2946-6-31
Comments
View archived comments (1)